This is “Mutations and Genetic Diseases”, section 19.5 from the book Introduction to Chemistry: General, Organic, and Biological (v. 1.0). For details on it (including licensing), click here.
For more information on the source of this book, or why it is available for free, please see the project's home page. You can browse or download additional books there. To download a .zip file containing this book to use offline, simply click here.



19.5 Mutations and Genetic Diseases
Learning Objective
- Describe the causes of genetic mutations and how they lead to genetic diseases.
We have seen that the sequence of nucleotides in a cell’s deoxyribonucleic acid (DNA) is what ultimately determines the sequence of amino acids in proteins made by the cell and thus is critical for the proper functioning of the cell. On rare occasions, however, the nucleotide sequence in DNA may be modified either spontaneously (by errors during replication, occurring approximately once for every 10 billion nucleotides) or from exposure to heat, radiation, or certain chemicals. Any chemical or physical change that alters the nucleotide sequence in DNA is called a mutationAny chemical or physical change that alters the nucleotide sequence in DNA.. When a mutation occurs in an egg or sperm cell that then produces a living organism, it will be inherited by all the offspring of that organism.
Common types of mutations include substitution (a different nucleotide is substituted), insertion (the addition of a new nucleotide), and deletion (the loss of a nucleotide). These changes within DNA are called point mutationsA change in which one nucleotide is substituted, added, or deleted. because only one nucleotide is substituted, added, or deleted (Figure 19.15 "Three Types of Point Mutations"). Because an insertion or deletion results in a frame-shift that changes the reading of subsequent codons and, therefore, alters the entire amino acid sequence that follows the mutation, insertions and deletions are usually more harmful than a substitution in which only a single amino acid is altered.
Figure 19.15 Three Types of Point Mutations
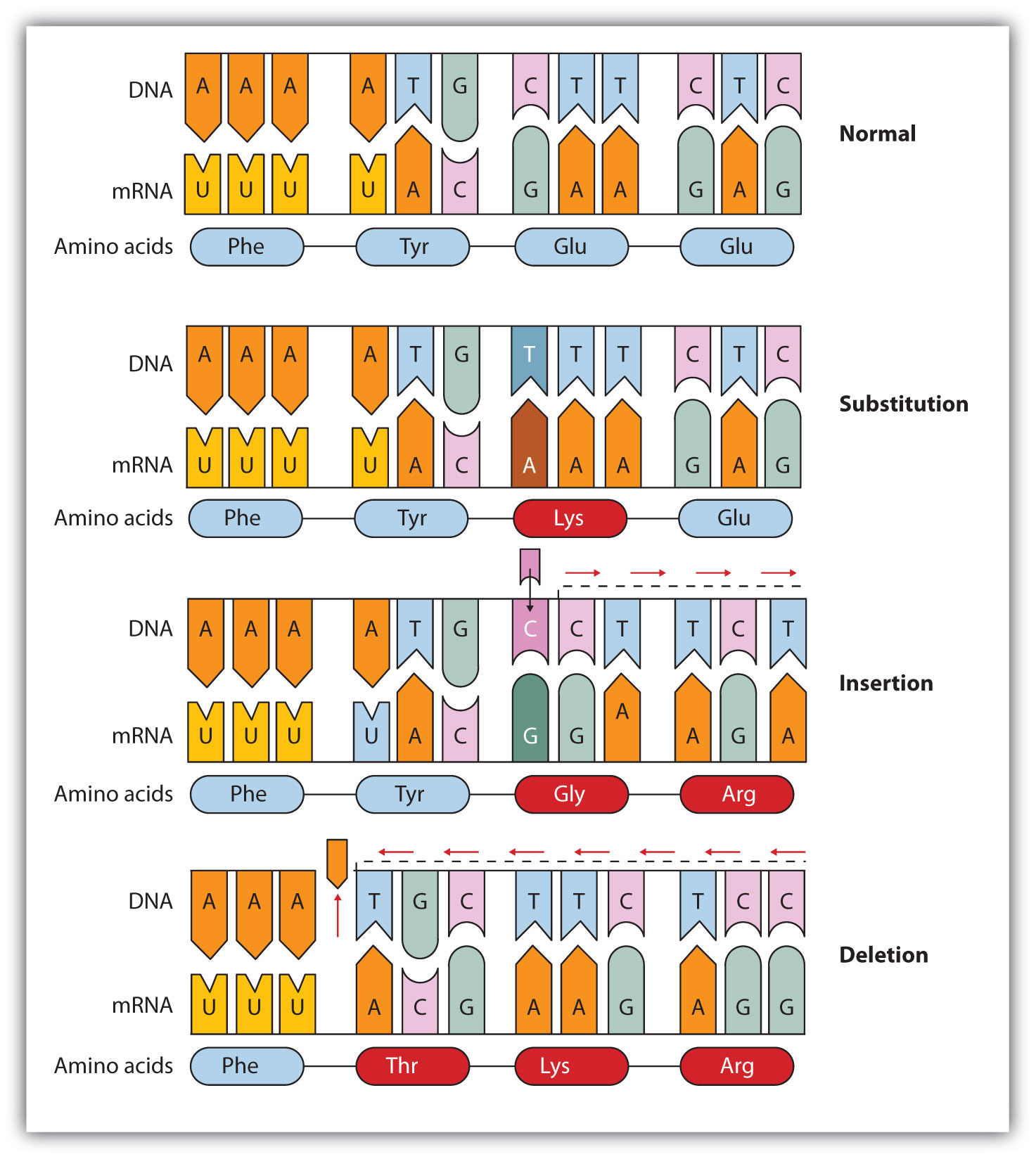
The chemical or physical agents that cause mutations are called mutagensA chemical or physical agent that cause mutations.. Examples of physical mutagens are ultraviolet (UV) and gamma radiation. Radiation exerts its mutagenic effect either directly or by creating free radicals that in turn have mutagenic effects. Radiation and free radicals can lead to the formation of bonds between nitrogenous bases in DNA. For example, exposure to UV light can result in the formation of a covalent bond between two adjacent thymines on a DNA strand, producing a thymine dimer (Figure 19.16 "An Example of Radiation Damage to DNA"). If not repaired, the dimer prevents the formation of the double helix at the point where it occurs. The genetic disease xeroderma pigmentosum is caused by a lack of the enzyme that cuts out the thymine dimers in damaged DNA. Individuals affected by this condition are abnormally sensitive to light and are more prone to skin cancer than normal individuals.
Figure 19.16 An Example of Radiation Damage to DNA
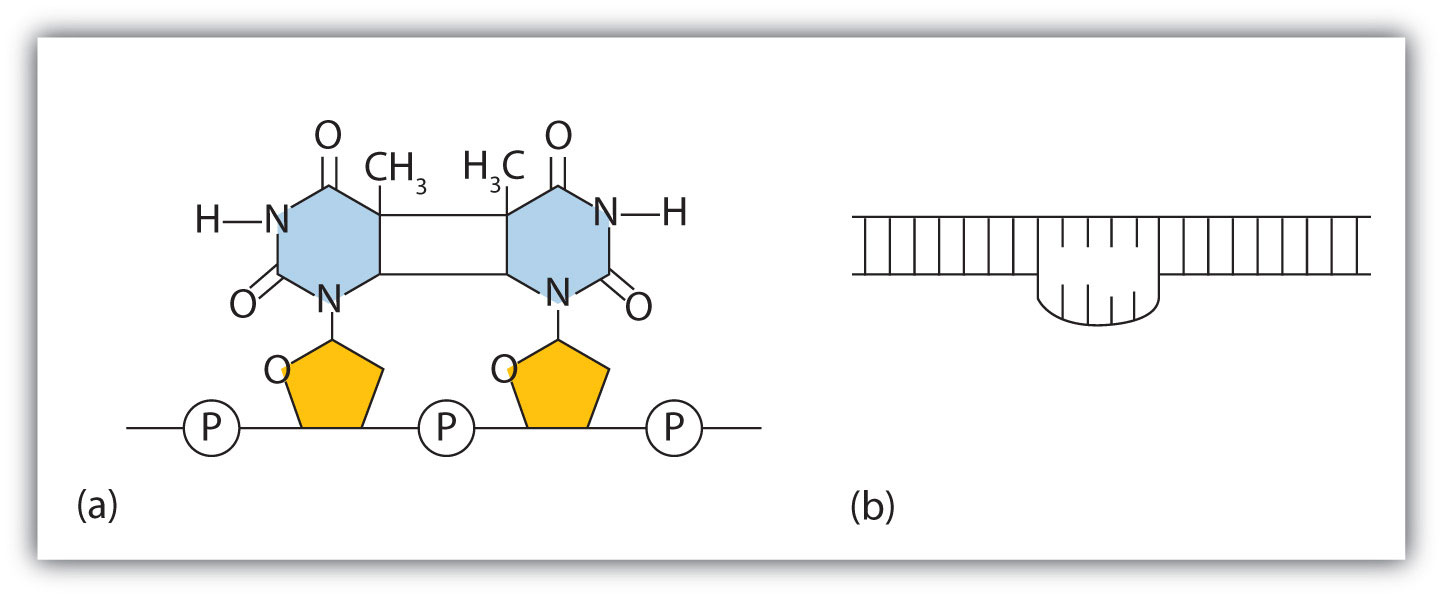
(a) The thymine dimer is formed by the action of UV light. (b) When a defect in the double strand is produced by the thymine dimer, this defect temporarily stops DNA replication, but the dimer can be removed, and the region can be repaired by an enzyme repair system.
Sometimes gene mutations are beneficial, but most of them are detrimental. For example, if a point mutation occurs at a crucial position in a DNA sequence, the affected protein will lack biological activity, perhaps resulting in the death of a cell. In such cases the altered DNA sequence is lost and will not be copied into daughter cells. Nonlethal mutations in an egg or sperm cell may lead to metabolic abnormalities or hereditary diseases. Such diseases are called inborn errors of metabolism or genetic diseasesA hereditary condition caused by an altered DNA sequence.. A partial listing of genetic diseases is presented in Table 19.3 "Some Representative Genetic Diseases in Humans and the Protein or Enzyme Responsible", and two specific diseases are discussed in the following sections. In most cases, the defective gene results in a failure to synthesize a particular enzyme.
Table 19.3 Some Representative Genetic Diseases in Humans and the Protein or Enzyme Responsible
| Disease | Responsible Protein or Enzyme |
|---|---|
| alkaptonuria | homogentisic acid oxidase |
| galactosemia | galactose 1-phosphate uridyl transferase, galactokinase, or UDP galactose epimerase |
| Gaucher disease | glucocerebrosidase |
| gout and Lesch-Nyhan syndrome | hypoxanthine-guanine phosphoribosyl transferase |
| hemophilia | antihemophilic factor (factor VIII) or Christmas factor (factor IX) |
| homocystinuria | cystathionine synthetase |
| maple syrup urine disease | branched chain α-keto acid dehydrogenase complex |
| McArdle syndrome | muscle phosphorylase |
| Niemann-Pick disease | sphingomyelinase |
| phenylketonuria (PKU) | phenylalanine hydroxylase |
| sickle cell anemia | hemoglobin |
| Tay-Sachs disease | hexosaminidase A |
| tyrosinemia | fumarylacetoacetate hydrolase or tyrosine aminotransferase |
| von Gierke disease | glucose 6-phosphatase |
| Wilson disease | Wilson disease protein |
PKU results from the absence of the enzyme phenylalanine hydroxylase. Without this enzyme, a person cannot convert phenylalanine to tyrosine, which is the precursor of the neurotransmitters dopamine and norepinephrine as well as the skin pigment melanin.
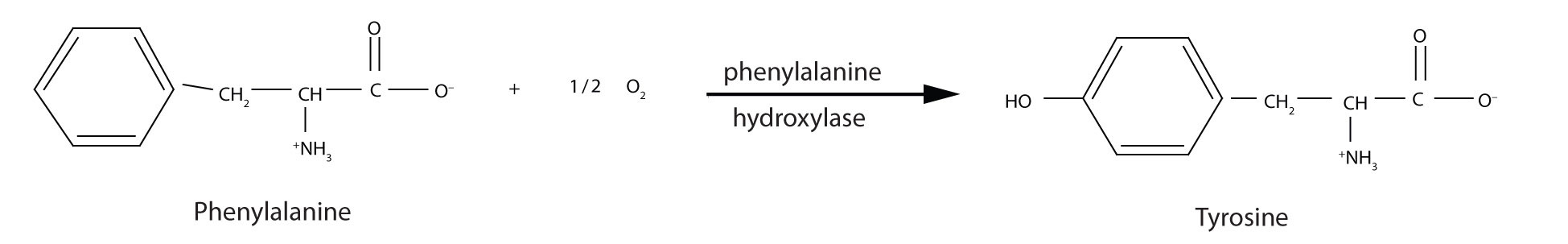
When this reaction cannot occur, phenylalanine accumulates and is then converted to higher than normal quantities of phenylpyruvate. The disease acquired its name from the high levels of phenylpyruvate (a phenyl ketone) in urine. Excessive amounts of phenylpyruvate impair normal brain development, which causes severe mental retardation.
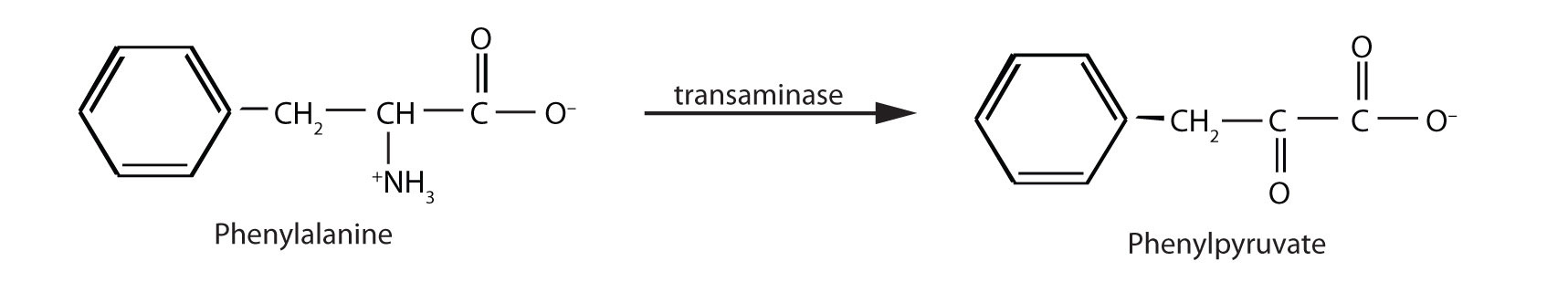
PKU may be diagnosed by assaying a sample of blood or urine for phenylalanine or one of its metabolites. Medical authorities recommend testing every newborn’s blood for phenylalanine within 24 h to 3 weeks after birth. If the condition is detected, mental retardation can be prevented by immediately placing the infant on a diet containing little or no phenylalanine. Because phenylalanine is plentiful in naturally produced proteins, the low-phenylalanine diet depends on a synthetic protein substitute plus very small measured amounts of naturally produced foods. Before dietary treatment was introduced in the early 1960s, severe mental retardation was a common outcome for children with PKU. Prior to the 1960s, 85% of patients with PKU had an intelligence quotient (IQ) less than 40, and 37% had IQ scores below 10. Since the introduction of dietary treatments, however, over 95% of children with PKU have developed normal or near-normal intelligence. The incidence of PKU in newborns is about 1 in 12,000 in North America.
Note
Every state has mandated that screening for PKU be provided to all newborns.
Several genetic diseases are collectively categorized as lipid-storage diseases. Lipids are constantly being synthesized and broken down in the body, so if the enzymes that catalyze lipid degradation are missing, the lipids tend to accumulate and cause a variety of medical problems. When a genetic mutation occurs in the gene for the enzyme hexosaminidase A, for example, gangliosides cannot be degraded but accumulate in brain tissue, causing the ganglion cells of the brain to become greatly enlarged and nonfunctional. (For more information on gangliosides, see Chapter 17 "Lipids", Section 17.3 "Membranes and Membrane Lipids".) This genetic disease, known as Tay-Sachs disease, leads to a regression in development, dementia, paralysis, and blindness, with death usually occurring before the age of three. There is currently no treatment, but Tay-Sachs disease can be diagnosed in a fetus by assaying the amniotic fluid (amniocentesis) for hexosaminidase A. A blood test can identify Tay-Sachs carriers—people who inherit a defective gene from only one rather than both parents—because they produce only half the normal amount of hexosaminidase A, although they do not exhibit symptoms of the disease.
Looking Closer: Recombinant DNA Technology
More than 3,000 human diseases have been shown to have a genetic component, caused or in some way modulated by the person’s genetic composition. Moreover, in the last decade or so, researchers have succeeded in identifying many of the genes and even mutations that are responsible for specific genetic diseases. Now scientists have found ways of identifying and isolating genes that have specific biological functions and placing those genes in another organism, such as a bacterium, which can be easily grown in culture. With these techniques, known as recombinant DNA technology, the ability to cure many serious genetic diseases appears to be within our grasp.
Isolating the specific gene or genes that cause a particular genetic disease is a monumental task. One reason for the difficulty is the enormous amount of a cell’s DNA, only a minute portion of which contains the gene sequence. Thus, the first task is to obtain smaller pieces of DNA that can be more easily handled. Fortunately, researchers are able to use restriction enzymes (also known as restriction endonucleases), discovered in 1970, which are enzymes that cut DNA at specific, known nucleotide sequences, yielding DNA fragments of shorter length. For example, the restriction enzyme EcoRI recognizes the nucleotide sequence shown here and cuts both DNA strands as indicated:
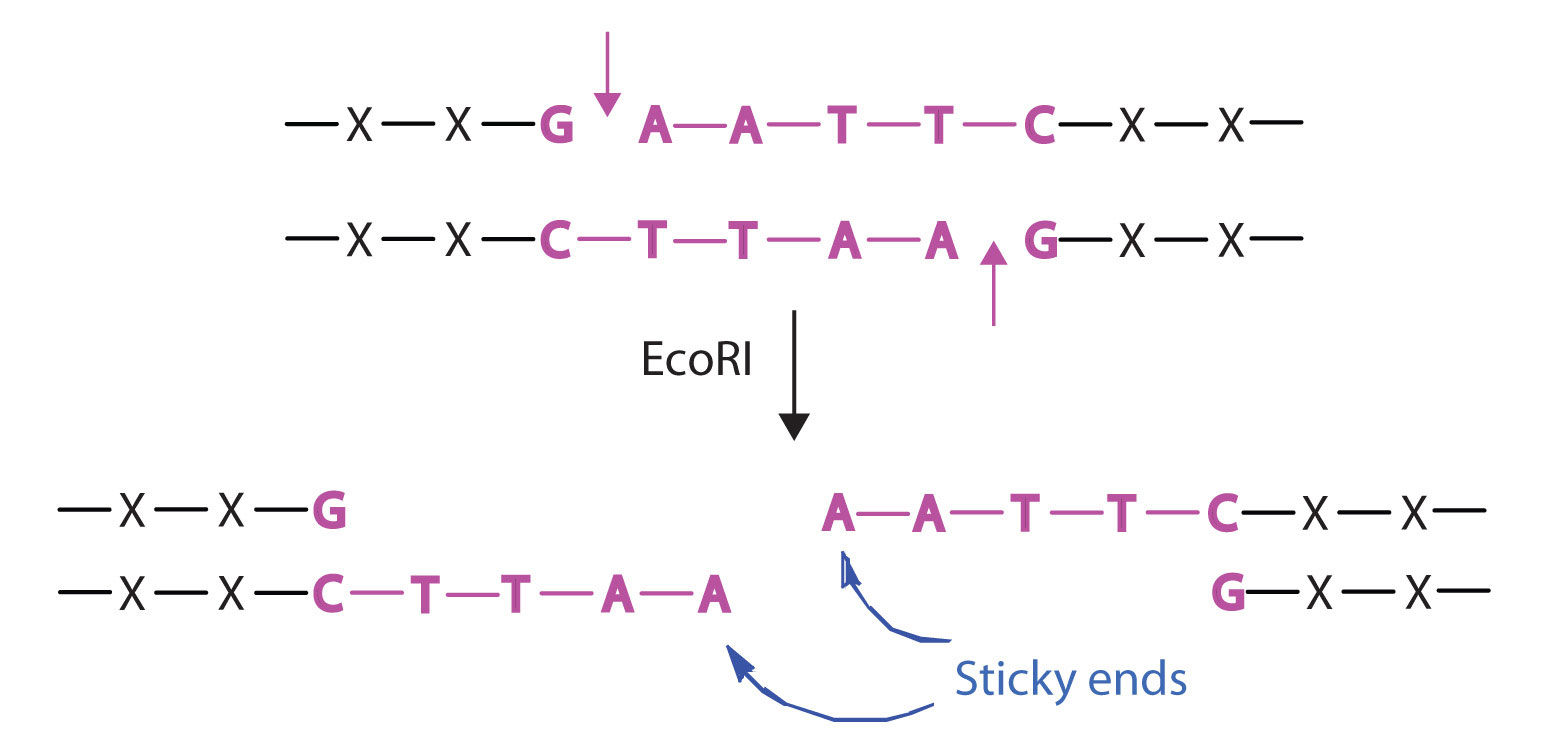
Once a DNA strand has been fragmented, it must be cloned; that is, multiple identical copies of each DNA fragment are produced to make sure there are sufficient amounts of each to detect and manipulate in the laboratory. Cloning is accomplished by inserting the individual DNA fragments into phages (bacterial viruses) that can enter bacterial cells and be replicated. When a bacterial cell infected by the modified phage is placed in an appropriate culture medium, it forms a colony of cells, all containing copies of the original DNA fragment. This technique is used to produce many bacterial colonies, each containing a different DNA fragment. The result is a DNA library, a collection of bacterial colonies that together contain the entire genome of a particular organism.
The next task is to screen the DNA library to determine which bacterial colony (or colonies) has incorporated the DNA fragment containing the desired gene. A short piece of DNA, known as a hybridization probe, which has a nucleotide sequence complementary to a known sequence in the gene, is synthesized, and a radioactive phosphate group is added to it as a “tag.” You might be wondering how researchers are able to prepare such a probe if the gene has not yet been isolated. One way is to use a segment of the desired gene isolated from another organism. An alternative method depends on knowing all or part of the amino acid sequence of the protein produced by the gene of interest: the amino acid sequence is used to produce an approximate genetic code for the gene, and this nucleotide sequence is then produced synthetically. (The amino acid sequence used is carefully chosen to include, if possible, many amino acids such as methionine and tryptophan, which have only a single codon each.)
After a probe identifies a colony containing the desired gene, the DNA fragment is clipped out, again using restriction enzymes, and spliced into another replicating entity, usually a plasmid. Plasmids are tiny mini-chromosomes found in many bacteria, such as Escherichia coli (E. coli). A recombined plasmid would then be inserted into the host organism (usually the bacterium E. coli), where it would go to work to produce the desired protein.
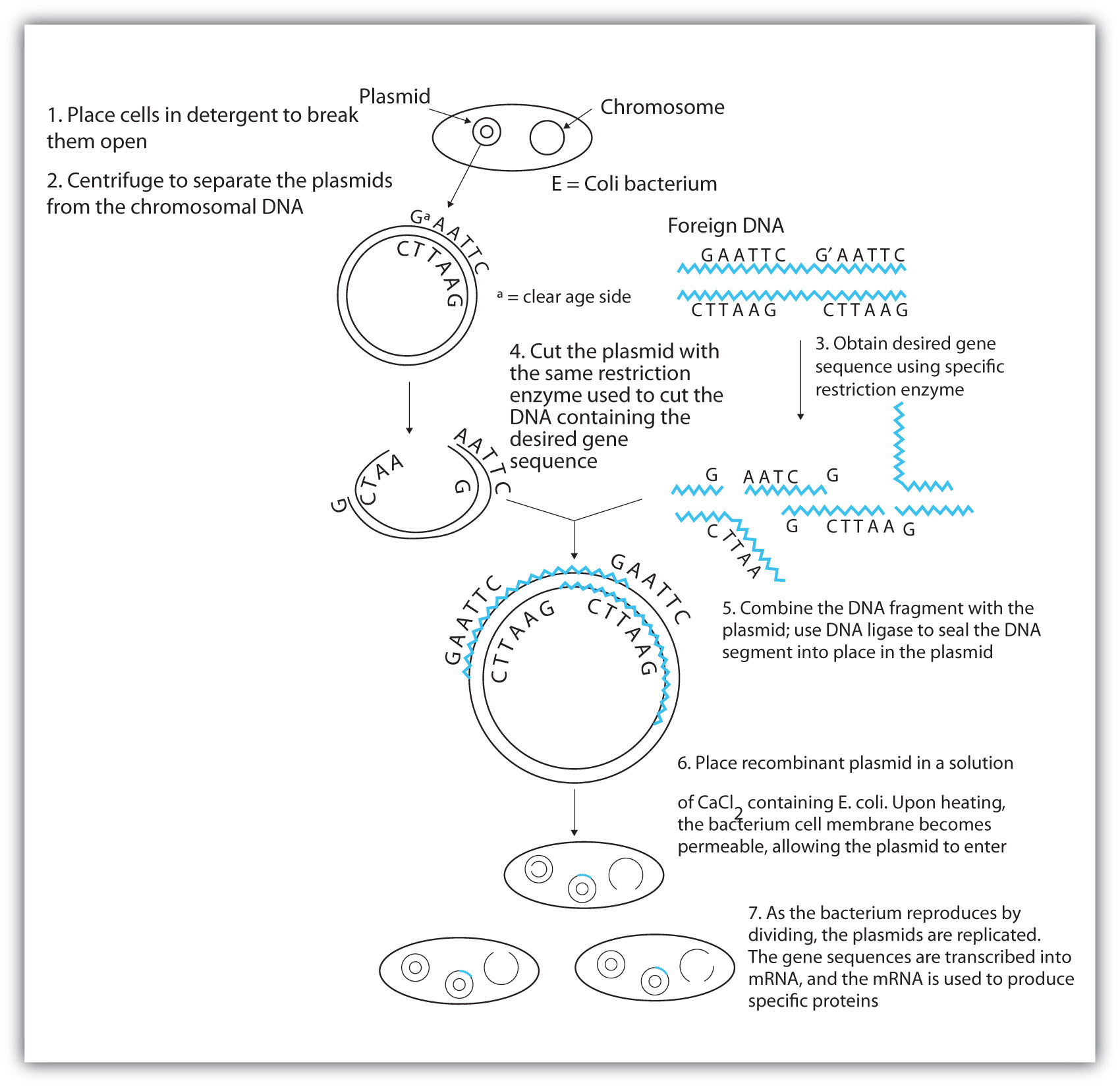
Proponents of recombinant DNA research are excited about its great potential benefits. An example is the production of human growth hormone, which is used to treat children who fail to grow properly. Formerly, human growth hormone was available only in tiny amounts obtained from cadavers. Now it is readily available through recombinant DNA technology. Another gene that has been cloned is the gene for epidermal growth factor, which stimulates the growth of skin cells and can be used to speed the healing of burns and other skin wounds. Recombinant techniques are also a powerful research tool, providing enormous aid to scientists as they map and sequence genes and determine the functions of different segments of an organism’s DNA.
In addition to advancements in the ongoing treatment of genetic diseases, recombinant DNA technology may actually lead to cures. When appropriate genes are successfully inserted into E. coli, the bacteria can become miniature pharmaceutical factories, producing great quantities of insulin for people with diabetes, clotting factor for people with hemophilia, missing enzymes, hormones, vitamins, antibodies, vaccines, and so on. Recent accomplishments include the production in E. coli of recombinant DNA molecules containing synthetic genes for tissue plasminogen activator, a clot-dissolving enzyme that can rescue heart attack victims, as well as the production of vaccines against hepatitis B (humans) and hoof-and-mouth disease (cattle).
Scientists have used other bacteria besides E. coli in gene-splicing experiments and also yeast and fungi. Plant molecular biologists use a bacterial plasmid to introduce genes for several foreign proteins (including animal proteins) into plants. The bacterium is Agrobacterium tumefaciens, which can cause tumors in many plants but which can be treated so that its tumor-causing ability is eliminated. One practical application of its plasmids would be to enhance a plant’s nutritional value by transferring into it the gene necessary for the synthesis of an amino acid in which the plant is normally deficient (for example, transferring the gene for methionine synthesis into pinto beans, which normally do not synthesize high levels of methionine).
Note
Restriction enzymes have been isolated from a number of bacteria and are named after the bacterium of origin. EcoRI is a restriction enzyme obtained from the R strain of E. coli. The roman numeral I indicates that it was the first restriction enzyme obtained from this strain of bacteria.
Concept Review Exercises
-
- What effect can UV radiation have on DNA?
- Is UV radiation an example of a physical mutagen or a chemical mutagen?
-
- What causes PKU?
- How is PKU detected and treated?
Answers
-
- It can lead to the formation of a covalent bond between two adjacent thymines on a DNA strand, producing a thymine dimer.
- physical mutagen
-
- the absence of the enzyme phenylalanine hydroxylase
- PKU is diagnosed by assaying a sample of blood or urine for phenylalanine or one of its metabolites; treatment calls for an individual to be placed on a diet containing little or no phenylalanine.
Key Takeaways
- The nucleotide sequence in DNA may be modified either spontaneously or from exposure to heat, radiation, or certain chemicals and can lead to mutations.
- Mutagens are the chemical or physical agents that cause mutations.
- Genetic diseases are hereditary diseases that occur because of a mutation in a critical gene.
Exercises
-
A portion of the coding strand of a gene was found to have the sequence 5′‑ATGAGCGACTTTCGCCCATTA‑3′. A mutation occurred in the gene, making the sequence 5′‑ATGAGCGACCTTCGCCCATTA‑3′.
- Identify the mutation as a substitution, an insertion, or a deletion.
- What effect would the mutation have on the amino acid sequence of the protein obtained from this mutated gene (use Figure 19.14 "The Genetic Code")?
-
A portion of the coding strand of a gene was found to have the sequence 5′‑ATGGCAATCCTCAAACGCTGT‑3′. A mutation occurred in the gene, making the sequence 5′‑ATGGCAATCCTCAACGCTGT‑3′.
- Identify the mutation as a substitution, an insertion, or a deletion.
- What effect would the mutation have on the amino acid sequence of the protein obtained from this mutated gene (use Figure 19.14 "The Genetic Code")?
-
- What is a mutagen?
- Give two examples of mutagens.
-
For each genetic disease, indicate which enzyme is lacking or defective and the characteristic symptoms of the disease.
- PKU
- Tay-Sachs disease
Answers
-
- substitution
- Phenylalanine (UUU) would be replaced with leucine (CUU).
-
-
- a chemical or physical agent that can cause a mutation
- UV radiation and gamma radiation (answers will vary)
-